

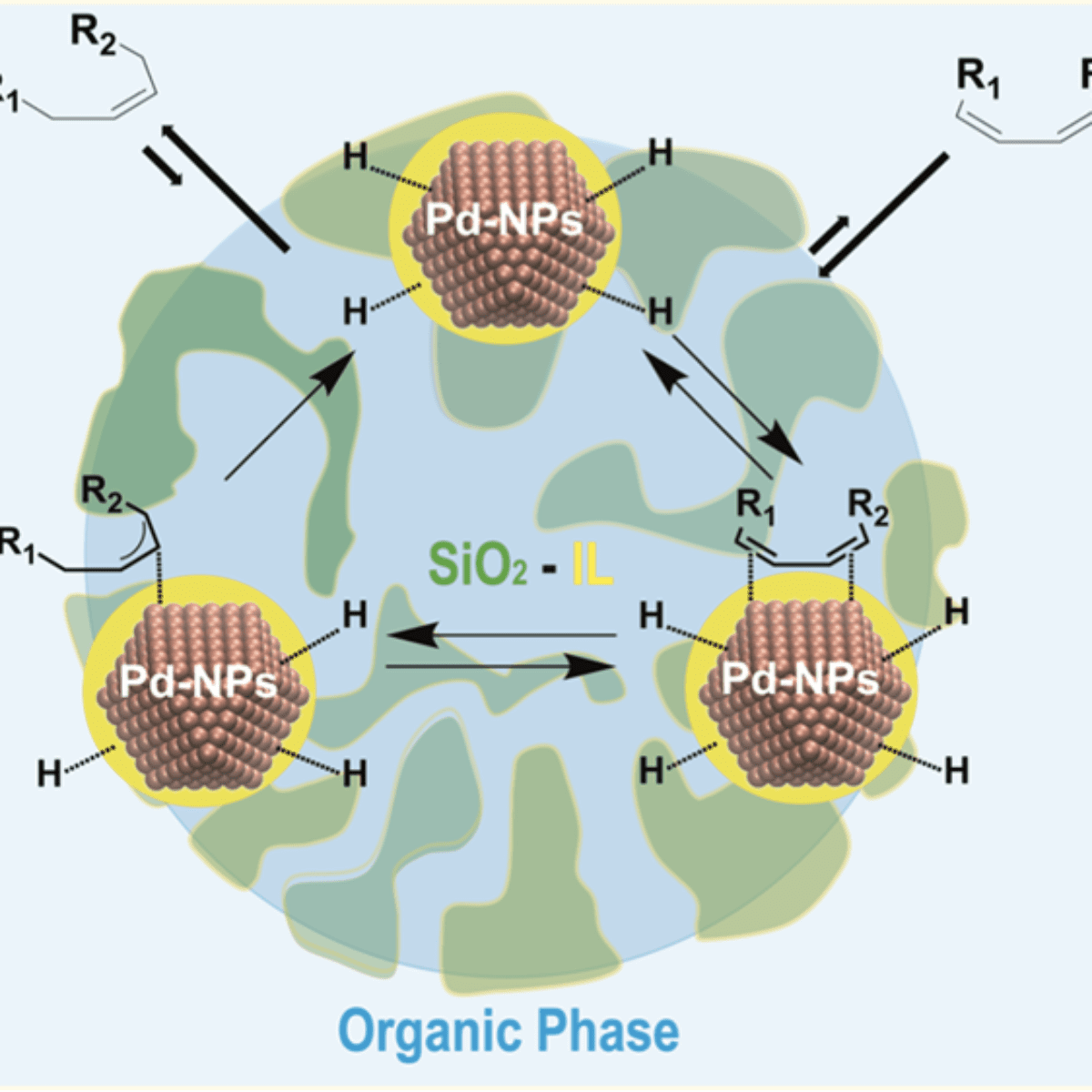
Results suggest that ionic liquid hybrid organosilica/Pd-NPs under multiphase conditions operate akin to catalytically active membranes.
It is well known that for many metal-catalyzed reactions the support influences the catalytic properties of the metal particles. The strong metal−support interaction (SMSI) effect may be due to: (i) geometrical effects: for example, the metallic nanoparticles (MNPs) are capped by functional groups from the support that migrate to the surface of the nanoparticles during the reaction; (ii) electronic effects: charge transfer between support and nanoparticle.
Indeed, the capping layer can create new catalytically active sites or also block access to them, the latter being detrimental for the catalytic properties of the nanoparticles.
However, the strong metal-support interaction effect is still not perfectly understood, especially for the case where the ionic liquid (IL) is covalently bonded to the support. The ionic liquid support effect can act as a physical barrier controlling the access or removal of reagents, intermediates, and products from the catalytically active site. This process looks like flowing liquid(s) (reagents and products) through a chemically active membrane composed of supported ionic liquid phase catalysts/NPs in confined space i.e., a non-equilibrium phenomenon (see Fig. 1). An important point of this non-equilibrium is that all the chemical transformations occurring with a catalyst in the course of its operation have to be considered as being inherently linked to the main “coupling” reaction. Indeed, the formation of ionic cages around organometallic complexes in thin films of supported ionic liquid phase catalysts has already been shown.
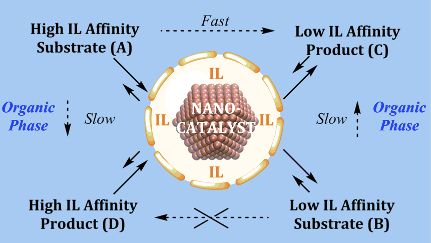
Figure 1: Schematic representation of immobilized catalysts in liquid ionic membrane-like device.
L. Luza et al., (1) have reported a strong catalytic support effect in palladium nanoparticles (Pd-NPs) supported on ionic liquid hybrid organosilicas. Apparently, the ionic liquid hydrophobicity plays a central role in the selective hydrogenation of dienes to monoenes (single double bond) by controlling the diene access to NP surface active sites.
More recently, L. Luza et al., (2) have tried to understand the nature of the metal−support interaction and to relate the support-induced changes in catalytic properties to the changes in electronic properties of the M-NPs. For this work, they have chosen hybrid organosilicas prepared by sol−gel processes using 1-n -butyl-3-(3-trimethoxysilylpropyl) imidazolium cations associated with hydrophilic and hydrophobic anions which can be easily decorated with well-dispersed and similar-sized (1.8− 2.1 nm) Pd-NPs by simple sputtering deposition. Moreover, these ionic liquid hybrid materials display solid membrane like properties.
The location of the sputter imprinted Pd-NPs on different supports, was determined by RBS (Rutherford backscattering) and HS-LEIS (low energy ion scattering) analysis and showed to be modulated by the strength of the contact ion pair formed between the imidazolium cation and the anion, rather than the ionic liquid hybrid organosilica pore size and surface area.
In contrast, the pore diameter and surface area of the hybrid supports display a direct correlation with the anion hydrophobicity. XPS (X-Ray Photoemission) analysis, done at LNLS, showed that the Pd (0) surface component decreases with increasing ionic bond strength between the imidazolium cation and the anions (contact ion pair). The finding is corroborated by changes in the coordination number associated with the Pd−Pd scattering in EXAFS measured at the LNLS XDS beamline. Hence, the interaction of the ionic liquid with the metal surface is found to occur via ionic liquid contact pairs (or aggregates). The observed selectivities of ≥ 99% to monoenes at full diene conversion indicate that the selectivity is intrinsic to the electron-deficient Pd metallic surfaces in this “restricted” ionic environment. This suggests that ionic liquid hybrid organosilica/Pd-NPs under multiphase conditions (“dynamic asymmetric mixture”) operate akin to catalytically active membranes: i.e., far from the thermodynamic equilibrium. Detailed kinetic investigations show that the reaction rate is zero order with respect to hydrogen and is dependent on the fraction of catalyst surfaces covered by either the substrate and/or the product. The reaction proceeds via rapid inclusion and sorption of the diene to the ionic liquid/Pd metal surface saturated with H species. This is followed by reversible hydride migration to generate a π-allyl intermediate. The reductive elimination of this intermediate, the formal rate-determining step, generates the alkene that is rapidly expelled from the ionic liquid phase to the organic phase.
Sources:
[1] L. Luza, A. Gual, C. P. Rambor, D. Eberhardt, S. R.Teixeira, F. Bernardi, D. L. Baptista and J. Dupont, Hydrophobic effects on supported ionic liquid phase Pd nanoparticle hydrogenation catalysts. J. Phys. Chem. Chem. Phys. 2014, 16, 18088−18091. DOI: 10.1039/C4CP03063J
[2] Leandro Luza, Camila P. Rambor, Aitor Gual, Fabiano Bernardi, Josiel B. Domingos, Thomas Grehl, Philipp Brüner, and Jairton Dupont, Catalytically Active Membranelike Devices: Ionic Liquid Hybrid Organosilicas Decorated with Palladium Nanoparticles. ACS Catal., 2016, 6 (10), pp 6478–6486. DOI: 10.1021/acscatal.6b01813
The proposed process could offer important technological and environmental advantages.

It is reported the magnetic characterization of the Fe3Ga4 intermetallic compound synthesized by the MFNN technique.